the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Apr 2019
| 04 Apr 2019
Technical note: Lithium isotopes in dolostone as a palaeo-environmental proxy – an experimental approach
Holly L. Taylor
Isaac J. Kell Duivestein
Juraj Farkas
Martin Dietzel
Anthony Dosseto
Lithium (Li) isotopes in marine carbonates have considerable potential as a proxy to constrain past changes in silicate weathering fluxes and improve our understanding of Earth's climate. To date the majority of Li isotope studies on marine carbonates have focussed on calcium carbonates. The determination of the Li isotope fractionation between dolomite and a dolomitizing fluid would allow us to extend investigations to deep times (i.e. Precambrian) when dolostones were the most abundant marine carbonate archives. Dolostones often contain a significant proportion of detrital silicate material, which dominates the Li budget; thus, pretreatment needs to be designed so that only the isotope composition of the carbonate-associated Li is measured. This study aims to serve two main goals: (1) to determine the Li isotope fractionation between Ca–Mg carbonates and solution, and (2) to develop a method for leaching the carbonate-associated Li out of dolostone while not affecting the Li contained within the detrital portion of the rock. We synthesized Ca–Mg carbonates at high temperatures (150 to 220 ∘C) and measured the Li isotope composition (δ7Li) of the precipitated solids and their respective reactive solutions. The relationship of the Li isotope fractionation factor with temperature was obtained:
Competitive nucleation and growth between dolomite and magnesite were observed during the experiments; however, there was no notable effect of their relative proportion on the apparent Li isotope fractionation. We found that Li isotope fractionation between the precipitated solid and solution is higher for Ca–Mg carbonates than for Ca carbonates. If the temperature of a precipitating solution is known or can be estimated independently, the above equation could be used in conjunction with the Li isotope composition of dolostones to derive the composition of the solution and hence make inferences about the past Li cycle. In addition, we also conducted leaching experiments on a Neoproterozoic dolostone and a Holocene coral. Results show that leaching with 0.05 M hydrochloric acid (HCl) or 0.5 % acetic acid (HAc) at room temperature for 60 min releases Li from the carbonate fraction without a significant contribution of Li from the siliciclastic detrital component. These experimental and analytical developments provide a basis for the use of Li isotopes in dolostones as a palaeo-environmental proxy, which will contribute to further advance our understanding of the evolution of Earth's surface environments.
- Article
(1676 KB) - Full-text XML
- BibTeX
- EndNote





Lithium isotopes in marine carbonates have emerged as a powerful proxy to help understand the evolution of the ocean chemistry, past silicate weathering fluxes and their links to global climate. Application to calcium carbonates (e.g. foraminifera, limestone) has shed some light on hotly debated topics such as, the evolution of Earth's climate during the Cenozoic (Misra and Froelich, 2012; Li et al., 2014; Wanner et al., 2014; Vigier and Goddéris, 2015; Hathorne and James, 2006), oceanic anoxic events (Pogge von Strandmann et al., 2013; Lechler et al., 2015) and Palaeozoic glaciation (Pogge von Strandmann et al., 2017). Although post-depositional alteration can play an important role in the formation of dolomite (Geske et al., 2012; Burns et al., 2000), the application of Li isotopes to marine dolostone could help to extend our understanding of the geochemical evolution of ancient dolomitizing solutions, particularly in early Earth geological history (i.e. Precambrian).
While data of Li isotopic fractionation during calcite precipitation have been relatively well constrained (Marriott et al., 2004a, b; Dellinger et al., 2018), there are currently no data available pertaining to Li isotope fractionation during dolomite formation. Therefore, in this study, precipitation experiments were carried out at various temperatures (150–220 ∘C), during which the Li isotopic composition of the precipitated solids and their respective reactive solutions were measured in order to determine the fractionation factor between the fluid and solid phases. The experiments were conducted at elevated temperatures due to the impossibility of synthesizing well-ordered dolomite at ambient temperatures on a laboratory timescale (Land, 1998; Arvidson and Mackenzie, 1999; Gregg et al., 2015).
One major difficulty with interpreting Li isotopes in dolostone is that they often contain a significant proportion of siliciclastic material (e.g. detrital micas and/or authigenic clay minerals). The abundance of Li in silicate minerals is higher than in carbonates (typically more than 2 orders of magnitude); thus, sample pretreatment must be undertaken to extract Li from only the carbonate fraction (Pogge von Strandmann et al., 2013; Bastian et al., 2018). Therefore, in this study we have tested various pretreatment methods in order to refine a procedure that faithfully yields the isotopic composition of the carbonate-associated Li fraction in dolostones exclusively.
2.1 Ca–Mg carbonate synthesis
Synthesis of Ca–Mg carbonates was conducted in Teflon-lined, stainless steel autoclaves at temperatures of 150, 180 and 220 ∘C ± 5 ∘C) through the reaction of ∼300 mg of powdered inorganic aragonite (speleothem aragonite; in-house mineral collection at Graz University of Technology) with an artificial brine solution containing 200 mM Mg, 0.245 mM Li and 50 mM NaHCO3. The reactive fluid was prepared by dissolving analytical grade MgCl2.6H2O (Roth; ≥99 %, p.a., ACS), LiCl (Merck; ≥99 %, ACS, Reag. Ph Eur) and NaHCO3 (Roth; ≥99.5 %, p.a., ACS, ISO) in ultrapure water (Millipore Integral 3: 18.2 MΩ cm−1). The stock solution was subsequently filtered through a 0.45 µm cellulose acetate membrane filter (Sartorius). The reagent inorganic aragonite was milled to a grain size <20 µm using a vibratory mill (McCrone Micronizing Mill) for 10 min and collected by dry sieving prior to use in the experiments. Autoclaves were sealed immediately after mixing the inorganic aragonite with the appropriate volume of stock solution and placed in preheated ovens. Samples were taken from the autoclaves at each operating temperature after a given reaction time (Table 1), including repeat samples. Upon removal from heat, the reactors were quenched and the samples were filtered through a 0.2 µm cellulose acetate membrane (Sartorius) using a vacuum filtration unit. Samples were then thoroughly rinsed with ultrapure water (Millipore Integral 3: 18.2 MΩ cm−1) to remove any soluble salts from the matrix and dried in an oven at 40 ∘C overnight to prepare them for subsequent solid phase analysis. An aliquot of the reactive fluid was acidified to a ∼3 % HNO3 matrix for elemental and Li isotope analyses using Merck® Suprapur™ HNO3.
Table 1Reaction temperatures and mineral content from the precipitation experiments.
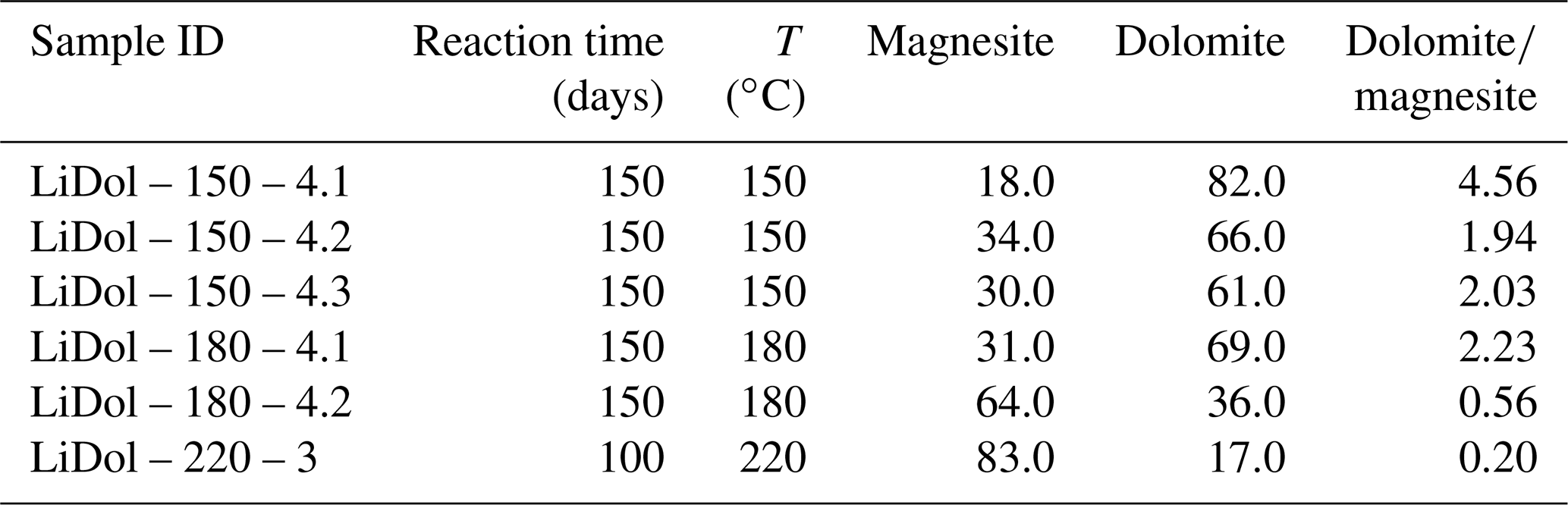
Mineral content in wt %. Note a maximum reaction time of 100 days was only possible at 220 ∘C, as no reacting solution was left after this time.
2.2 Leaching experiments
A Neoproterozoic dolostone from the Nuccaleena Formation (Flinders Ranges, South Australia, ∼635 Ma) and a Holocene Porites coral were used to evaluate the effect of different leaching protocols on the measured Li isotope composition. Samples were ground to a powder using a TEMA chromium-ring grinding mill. An aliquot of powdered dolostone was used for mineralogy quantification performed using X-ray diffraction. Another aliquot of 1 g was placed in a clean polypropylene centrifuge tube and 20 mL of solution was added. Leaching was tested with hydrochloric acid (HCl) of varying concentrations (0.05, 0.1, 0.15, 0.2, 0.3, 0.5, 0.8, 1 and 6) and acetic acid (HAc) at concentrations of 0.5 % and 2 %. HAc and HCl solutions were prepared from trace grade glacial acetic acid (Merck® Suprapur™) and ultra-trace grade 30 % HCl (Merck® Suprapur™). In each case, the powder and solution reacted at room temperature for 1 h, while continuous mixing was achieved with an orbital shaker. The supernatant fluid was separated by centrifugation at 4000 rpm for 15 min. After separation, the supernatant fluid was extracted using acid-washed disposable pipettes. An aliquot containing ∼60 ng of Li was subsequently sampled for cation exchange chromatography.
2.3 Mineralogy quantification
Quantitative phase contents of the synthesized solids were determined by powder X-ray diffraction (XRD) of finely ground aliquots performed on a PANalytical X'Pert PRO diffractometer outfitted with a Co-target tube (operated at 40 kV and 40 mA), a high-speed X'Celerator Scientific detector, 0.5∘ antiscatter and divergence slits, spinner stage, primary and secondary Soller and automatic sample changer. Samples were finely ground by hand using a mortar and pestle prior to analysis and were loaded in a random orientation using the top loading technique. The samples were analysed over the 4–85∘2θ range with a step size of 0.008∘2θ and a count time of 40 s step−1. Mineral quantification was obtained by Rietveld refinement of the XRD patterns using the PANalytical X'Pert HighScore Plus software and its implemented pdf-2 database.
2.4 Elemental concentrations
Lithium concentrations of solutions were analysed in acidified (0.3 M HNO3) aliquots by inductively coupled plasma optical emission spectroscopy (ICP-OES) using a PerkinElmer Optima 8300. A range of in-house and NIST 1640a standards were measured at the beginning and end of a sample series, with an estimated analytical error (2σ, three replicates) of ±3 % relative to the standard. For synthesized solids, an aliquot of each precipitate was dissolved in 0.9 M HNO3 at 70 ∘C for 12 h in an ultrasonic bath to ensure complete digestion. Subsequently, Li concentrations were analysed by ICP-OES following the same method as for the aqueous solutions.
2.5 Lithium isotopes
Sample preparation for Li isotope measurement was undertaken in a class 100 clean room at the Wollongong Isotope Geochronology Laboratory, University of Wollongong. For mineral precipitates, the samples were ground using a mortar and pestle before aliquots of <0.05 g were weighed. The sample aliquots were dissolved in dilute HNO3 (Ultrapur™), and 0.2 mL of concentrated H2O2 (31 % Ultrapur™) was added to ensure the breakdown of organics.
The samples were then placed on a hotplate overnight at 50 ∘C to reflux and ensure complete digestion of the solids. After complete digestion of the solids, Li concentrations were measured by quadrupole ICP-MS. An aliquot of the digested samples containing ∼60 ng of Li was then dried down and taken up into 1.5 mL of Ultrapur™ 1 M HCl. Samples were then treated with a two-step cation exchange chromatography procedure, following the methods of Balter and Vigier (2014) to separate Li from the sample matrix. For Li isotope measurements it is crucial that 100 % of Li is recovered from the cation exchange columns as δ7Li compositions have been shown to vary by up to ∼200 ‰ during chromatography due to incomplete recovery (Pistiner and Henderson, 2003). It is also crucial to remove elements such as Na and Ca as large amounts of Ca can coat the cones of the mass spectrometer, whereas Na can reduce Li ionization in the plasma and cause further Li isotopic fractionation during analysis (James and Palmer, 2000). For chromatography, 30 mL Savillex micro columns (6.4 mm internal diameter, 9.6 cm external diameter, 25 cm capillary length) were used together with Bio-Rad AG50W-X8 resin as the cation exchange medium (volume of 3.06 cm3). The columns were calibrated with seawater prior to treating the samples to verify that the procedure yielded 100 % of the Li (Table A1 in the Appendix). The columns were cleaned with 30 mL of 6M HCl, rinsed with 2 mL of MilliQ™ water and conditioned using 8 mL of titrated, 1 M Ultrapur™ HCl before sample loading. To ensure the complete removal of interfering elements from the Li, samples were passed through the columns twice; after the first elution, the samples were dried down, taken up in 1 M HCl and reloaded into the columns. The Li elutions were dried down and subsequently re-dissolved in Ultrapur™ 0.3 M HNO3 ready for isotopic analysis. Lithium isotope ratios were measured by multi-collector inductively coupled plasma mass spectrometry (MC ICP-MS) on a ThermoFisher Neptune Plus at the Wollongong Isotope Geochronology Laboratory, University of Wollongong. A 30 ppb solution of IRMM-16 Li isotopic standard was used at the start of each measurement session to tune the instrument. An intensity of ∼1 V was routinely obtained for 7Li, whereas the background 7Li intensity was between 5 and 30 mV. During analysis, standard bracketing, using IRMM-16 as the primary standard, was applied to correct the measured 7Li∕6Li values for mass bias (Flesch et al., 1973). Instrumental blanks were measured before each sample so that the background signal could be accounted for. The 7Li∕6Li ratios were converted to δ7Li values using L-SVEC as reference to Carignan et al. (2007) (Eq. 1).
The accuracy of the analysis was assessed using synthetic solutions, Li6-N and Li7-N (Carignan et al., 2007), as secondary standards every six samples. The accuracy of the chromatography and analysis was assessed using a seawater standard (Table A1). External uncertainty on δ7Li compositions (at the 2 σ level) was evaluated by measurement of precipitated solids and solutions from repeat experiments at 150 ∘C (n=3) and 180 ∘C (n=2), and amounted to 0.86 ‰ for precipitated solids and 1.2 ‰ for solutions.
3.1 Precipitation experiments
Synthesized minerals are comprised of dolomite and magnesite (Table 1); their relative amount shows a relationship with temperature, with higher reaction temperatures yielding more magnesite and less dolomite compared with lower temperatures (Fig. 1). The Li concentration of reactive solutions ranges from 1666 to 3695 µg L−1 (Table A2) and shows no correlation with reaction temperature. On the contrary, the Li concentration of precipitated solids is consistent with (Marriott et al., 2004a, b) as it decreases with increasing temperature (from 25.9 to 8.20 ppm; Table A2).
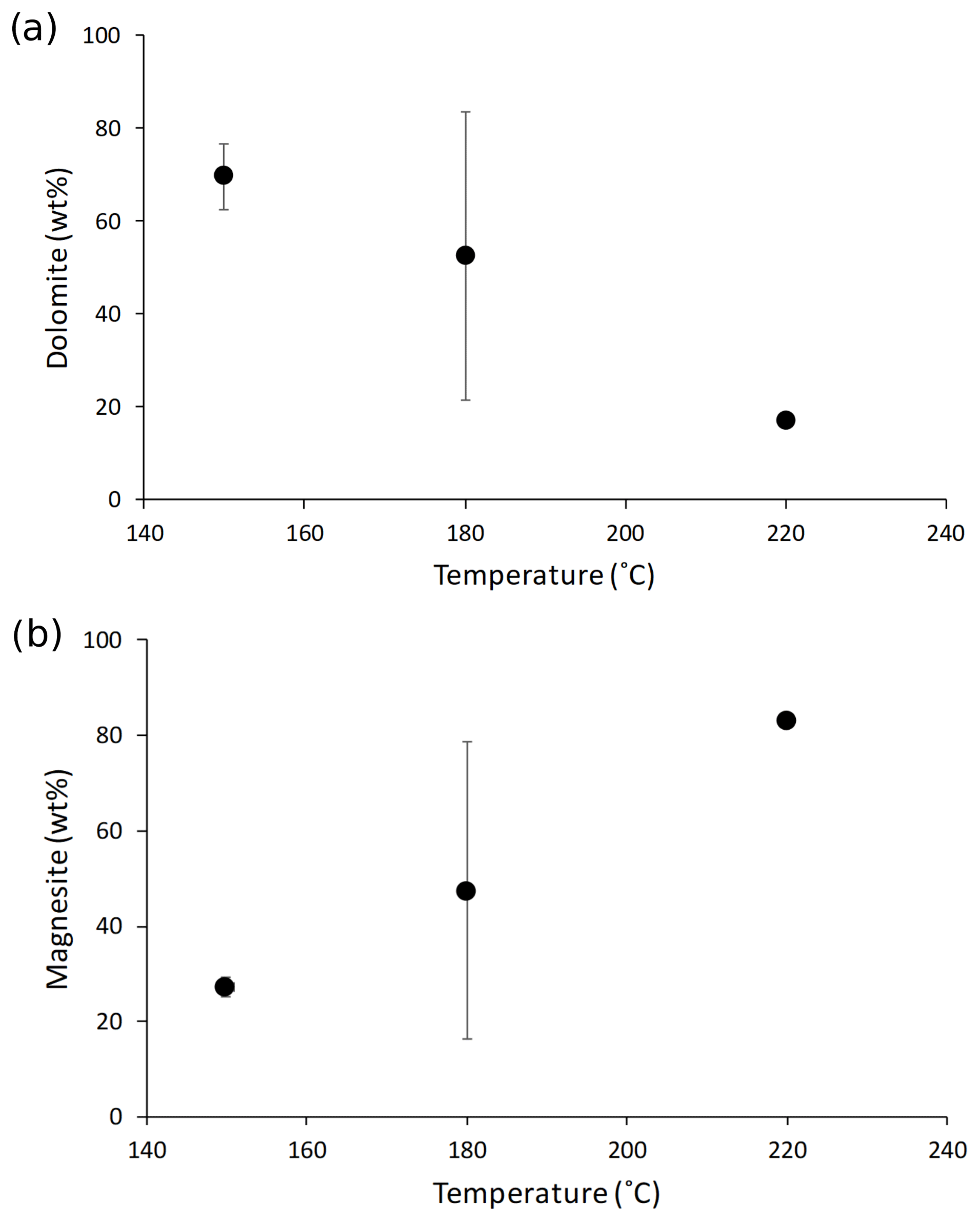
Figure 1(a) Dolomite and (b) magnesite concentrations in the precipitated solid (in wt %) as a function of reaction temperature (in ∘C). The data displayed are average values for each reaction temperature. The error is not shown for mineral concentrations at 220 ∘C because no repeat analysis was performed. The error on the magnesite content at 150 ∘C is within the symbol size.
The δ7Li of the initial reactive solution is 7.85 ‰ (Table 2). After reaction the δ7Li values of the solution (δ7Lisol) vary between 7.87 and 9.48 ‰, whereas the δ7Li values in the precipitated solid (δ7Liprec) range from −0.63 ‰ to 3.08 ‰ (Table 2). The precipitated solids are 4.79 ‰ to 8.6 ‰ lighter than the solution, and this difference (termed 103ln αprec-sol) increases with decreasing temperature (Table 2).
Table 2Li isotope composition in solutions and precipitated solids for the precipitation experiments.

External uncertainty (at the 2 σ level) is 0.86 ‰ and 1.2 ‰ on the δ7Li values of precipitated solids and solutions, respectively.
The Li isotope fractionation factors of the precipitated solid and the solution, calculated as , display values within error of each other, despite the wide range of concentrations of dolomite or magnesite precipitated (Fig. 2). Similarly, there is no relationship between the Li distribution coefficient between precipitated solid and solution (, where [Li]prec and [Li]sol are the Li concentrations in the precipitated solid and the solution, respectively), and mineral abundances (Fig. 3). Conversely, there is a positive relationship between 103ln αprec-sol and the reaction temperature (Fig. 4).
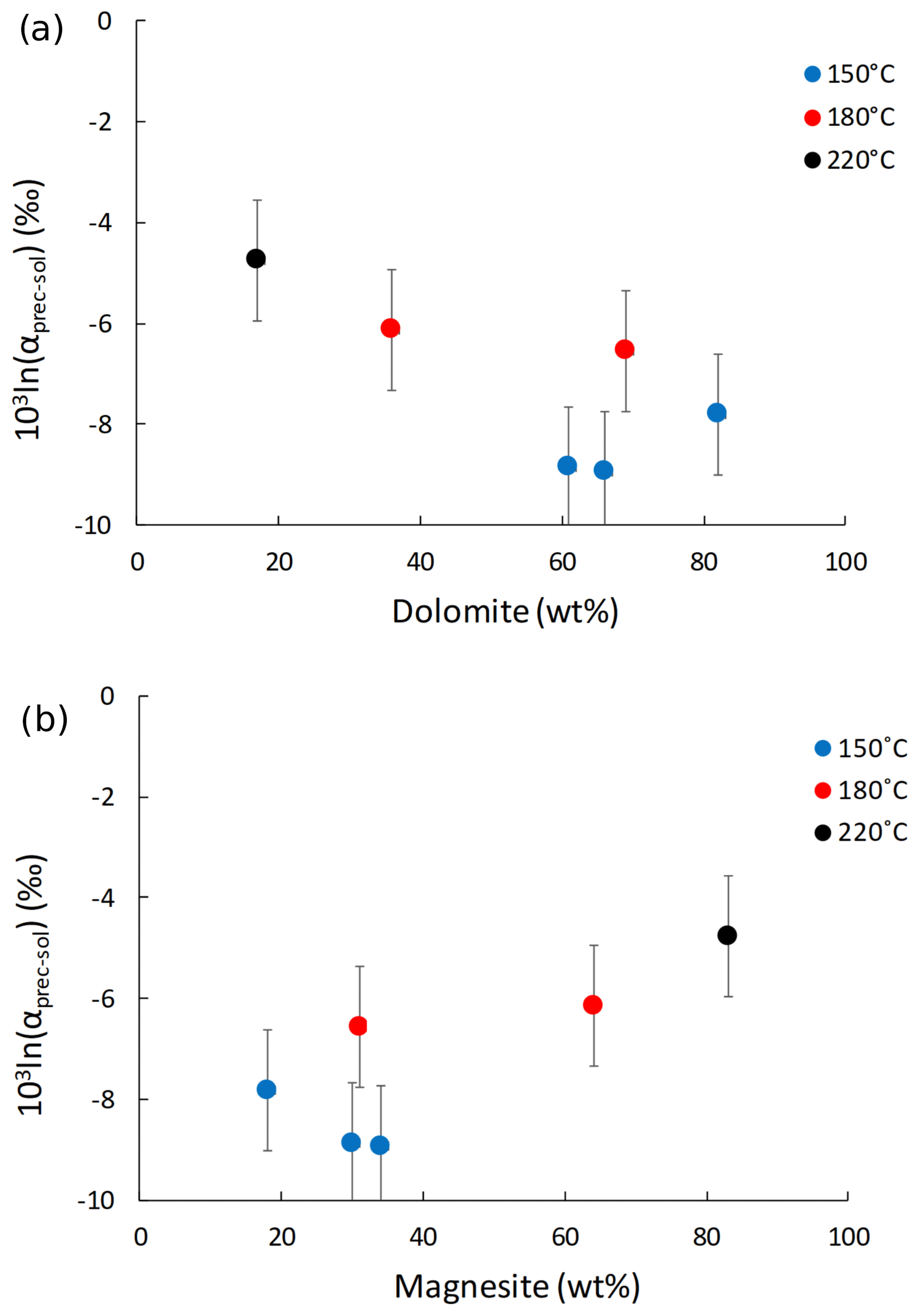
Figure 2Lithium isotope fractionation factor between the precipitated solid and the solution (103ln αprec-sol) as a function of (a) dolomite and (b) magnesite content (in wt %).

Figure 3The distribution coefficient of Li between solid and solution (D[Li]prec-sol) as a function of (a) dolomite content and (b) magnesite content (in wt %).
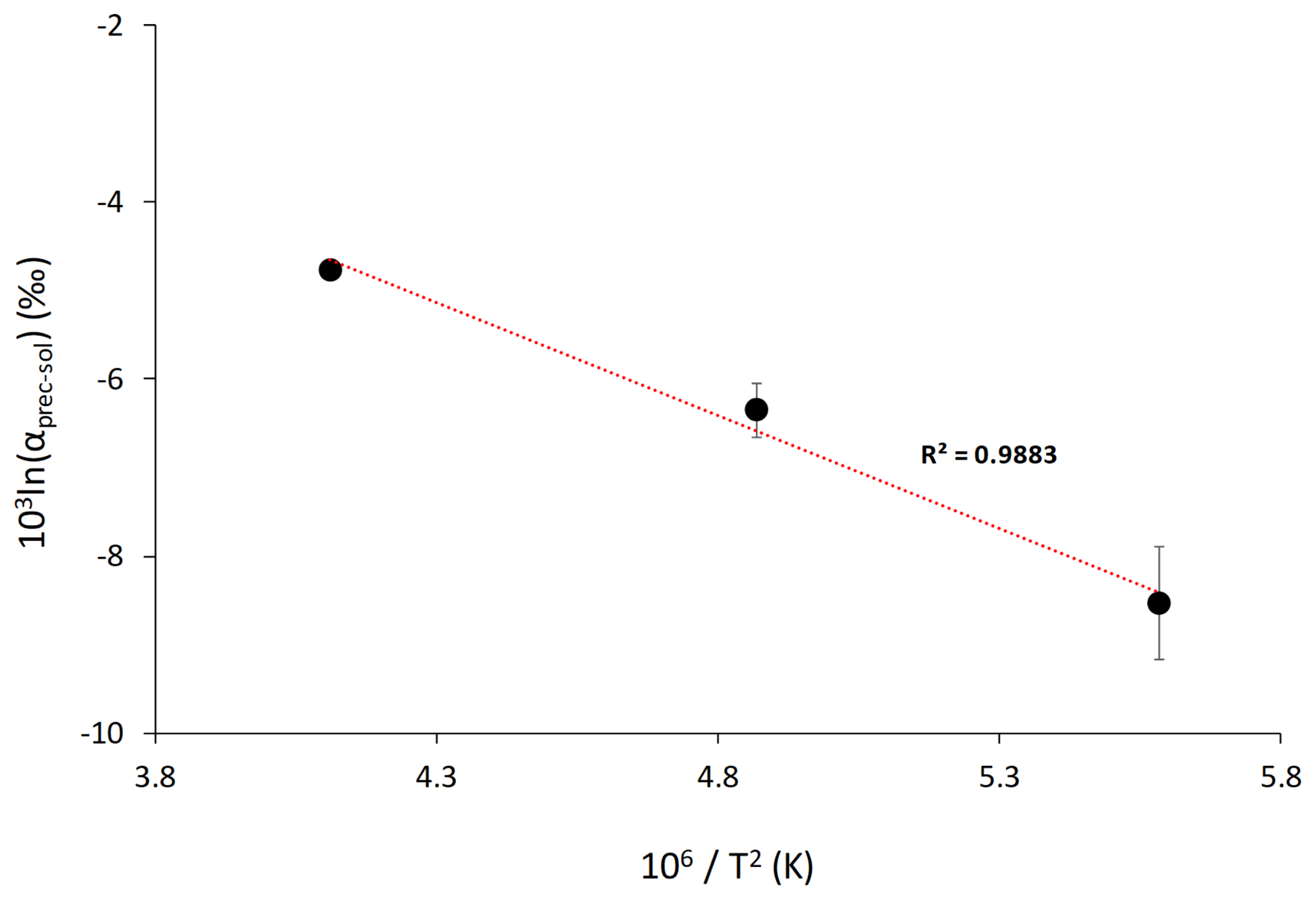
Figure 4Lithium isotope fractionation factor as a function of the reaction temperature, T (in K). Average values for each temperature are shown. The dotted line shows the linear regression through these values according to Eq. (2). Error is not shown for the isotope fractionation factor at 220 ∘C because no repeat analysis was performed.
3.2 Leaching experiments
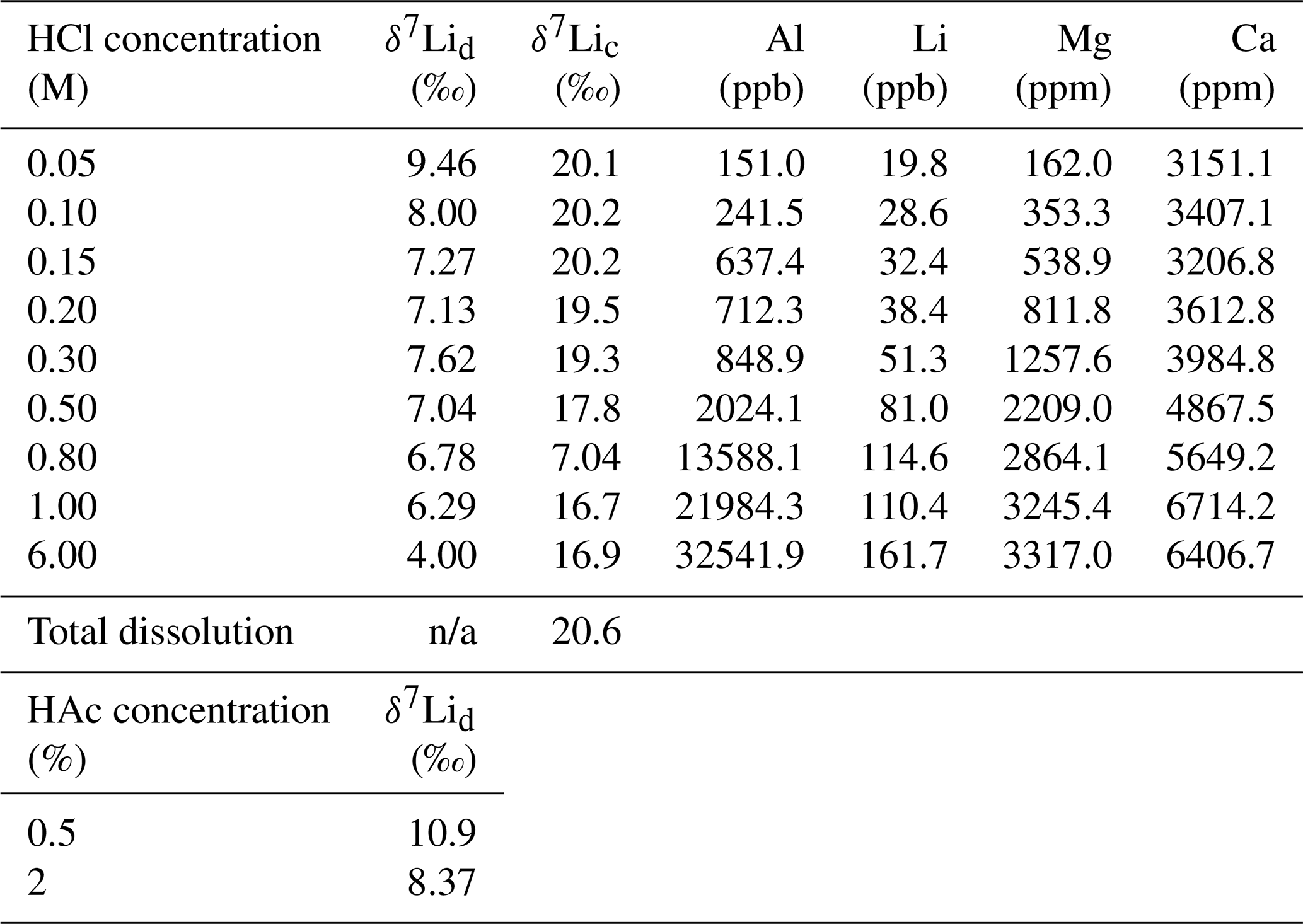
For the dolostone, δ7Li values of the leaching solution decrease from 9.5 to 4.0 ‰, with increasing HCl concentration (Table 3; Fig. 5a). The molar Al∕Mg ratio in the leaching solutions increases at HCl concentrations >0.8 M from ∼0.0009 to 0.01 (Fig. 5b). The leaching solutions show an increase in the molar Li∕Ca ratio from to with decreasing δ7Li (Fig. 6a). Furthermore, the molar Li∕Mg ratio increases from 5 to with increasing δ7Li (Fig. 6b). Very few carbonate minerals other than dolomite (1.1 wt % calcite and 2.1 wt % ankerite) are present in the dolostone sample, and the silicate minerals represents ∼26 wt % of the sample (14 wt % quartz, 6.2 wt % muscovite and 5.1 wt % albite) (Table A3). Leaching with HAc yields δ7Li compositions in the solution similar to values observed with very dilute HCl (Fig. 7). The δ7Li of the 2 % HAc leaching solution is lower than that of the 0.5 % HAc leaching solution.
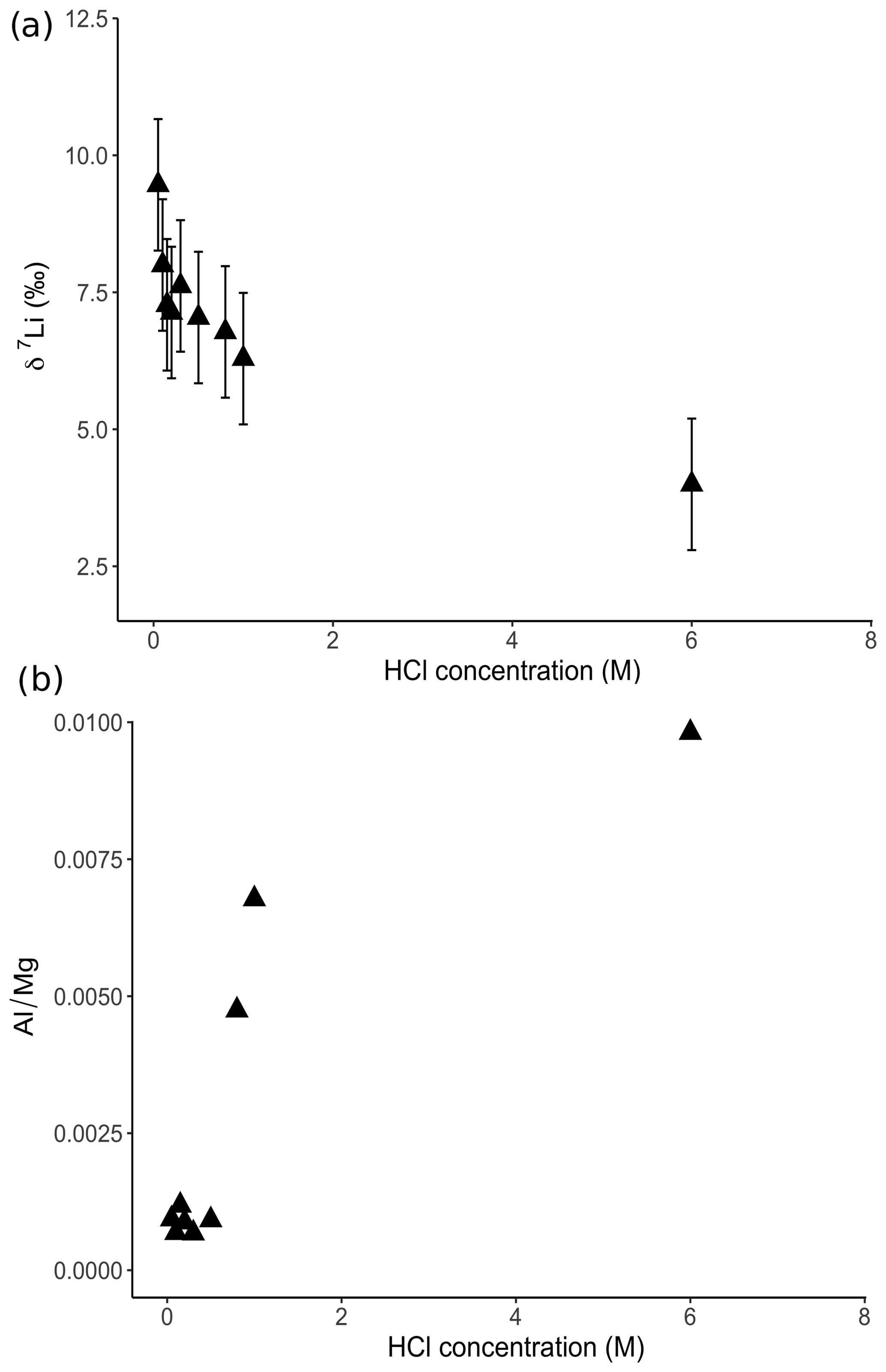
Figure 5(a) Lithium isotope compositions and (b) Al∕Mg ratios of solutions from dolostone leaching, as a function of their HCl concentration. Decreasing δ7Li values with increasing HCl concentration suggest a release of isotopically light Li from clay minerals, which is supported by the increase in the Al∕Mg ratios. Error bars are within the symbol size, if not shown.
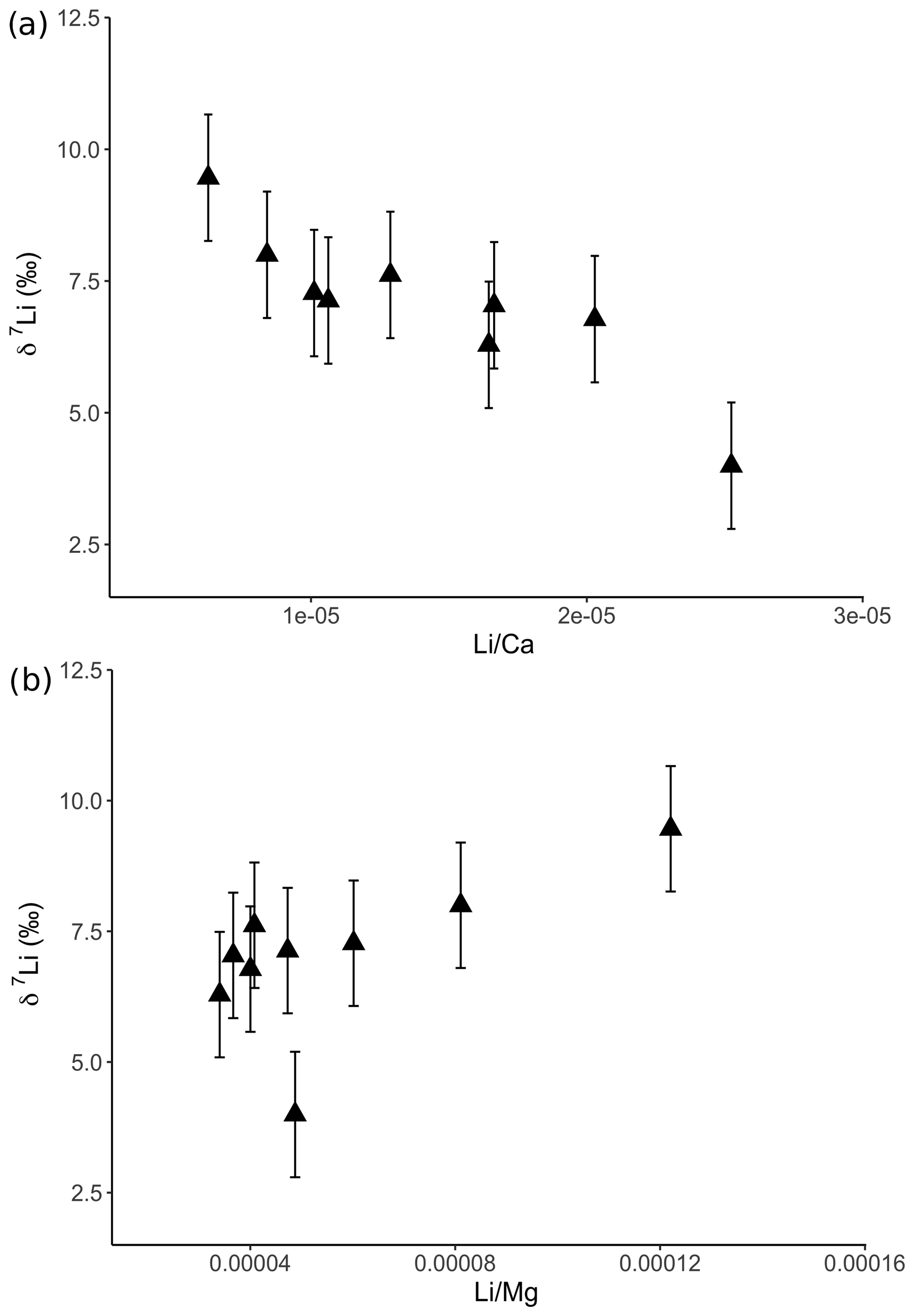
Figure 6Lithium isotopic compositions of solutions from dolostone leaching, as a function of their (a) Li∕Ca and (b) Li∕Mg ratios. Error bars are within the symbol size, if not shown.
For the Holocene coral, the sample is dominated by aragonite (Table A4) and the leaching solution shows a similar trend to that from the dolostone leaching experiment, with δ7Li values decreasing from 20.1 to 16.9 with increasing HCl concentration (Table 3; Fig. 8). Total dissolution of the coral yields a δ7Li value in the solution of 20.6 ‰, which is within error of the values determined for HCl leaching experiments with acid concentrations <0.5 M (Table 3).
4.1 Lithium isotope fractionation during inorganic precipitation of Ca–Mg carbonate
The precipitated solids of the synthesis experiments consist of Ca–Mg carbonates with variable amounts of dolomite (CaMg(CO3)2) and magnesite (MgCO3) (Table 1). The δ7Li composition of the precipitated solids is systematically isotopically lighter than that of the reactive solution (Table 2). These results are consistent with previous experimental work on Li isotope fractionation during calcite precipitation (Marriott et al., 2004a, b), which showed that the Li isotope composition of calcite is isotopically lighter than that of the corresponding fluid. Teng et al. (2008) suggested that the incorporation of 6Li over 7Li in minerals compared to the growth solution reflects a change from four- to six-fold coordination of Li during mineral growth. In calcite from foraminifera and aragonite from corals, δ7Li values are about 3 ‰ and 11 ‰ lower, respectively, compared with their growth solutions (Marriott et al., 2004a). Here, the precipitated minerals are 4.8 ‰ to 8.6 ± 0.6 ‰ (1σ; n=3) lighter than the solution over all temperatures (150, 180 and 220 ∘C). This difference increases with decreasing temperature, as would be expected for stable isotope fractionation at equilibrium. As our experiments were conducted at high temperatures (150, 180 and 220 ∘C), the system can reasonably be considered to be approaching isotope equilibrium conditions as fractionation scales with the inverse of reaction temperature (see Fig. 3). Marriott et al. (2004a) suggested that Li isotope fractionation probably occurs at equilibrium even at lower temperatures for several reasons: (i) kinetic fractionation would probably be much greater (up to ∼80 ‰) than that observed (both in calcite and in Ca–Mg carbonate), and would therefore require boundary layer processes or the presence of a back-reaction, for which there is no evidence; (ii) observed isotopic fractionation between calcite and growth solution, as well as between Ca–Mg carbonate and growth solution, are consistent with ab initio calculations for equilibrium fractionation (Kazuyo et al., 2001); and (iii) lithium isotope fractionation between calcite and growth solution is relatively constant across a wide range of concentrations of Li incorporated in calcite (this was not tested here).
Table 3Lithium isotope compositions and elemental concentrations of solutions from the dolostone and coral leaching experiments with HCl and HAc.
δ7Lid (‰) and δ7Lic (‰) are the Li isotope composition of solutions from dolostone and coral leaching experiments, respectively. n/a denotes not applicable.
Although Li isotope fractionation and the magnesite∕dolomite ratio of the precipitated solid both co-vary with temperature, there is no relationship between the δ7Li composition of the precipitated solids or that of their respective reactive solutions and the magnesite∕dolomite ratio of the precipitated solid (not shown). This suggests that the nature of the Ca–Mg carbonate precipitated does not have a significant influence on Li isotope fractionation. This hypothesis is supported by the absence of significant variation in the Li isotope fractionation factor (103ln αprec-sol; Fig. 2) or the Li distribution coefficient between solid and solution (D[Li]prec-sol; Fig. 3), despite a wide range of mineral abundances. For instance, most 103ln αprec-sol values are within error of each other, whereas the dolomite concentration varies from 17 wt % to 82 wt % (Fig. 2a). This differs from what Marriott et al. (2004b) observed for calcium carbonates at ambient temperature, where the isotopic fractionation in aragonite (∼11 ‰) was much greater than in calcite (∼3 ‰).
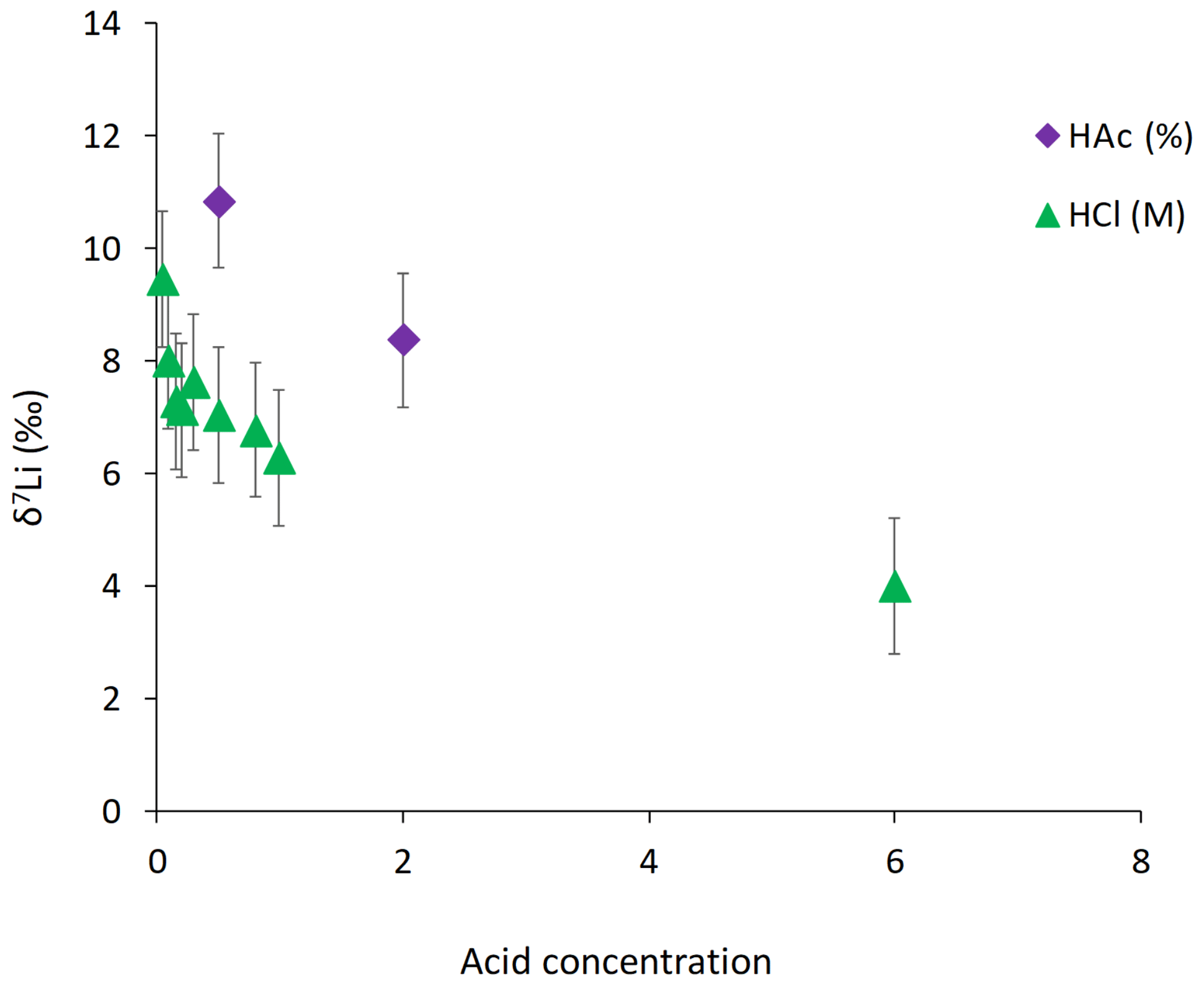
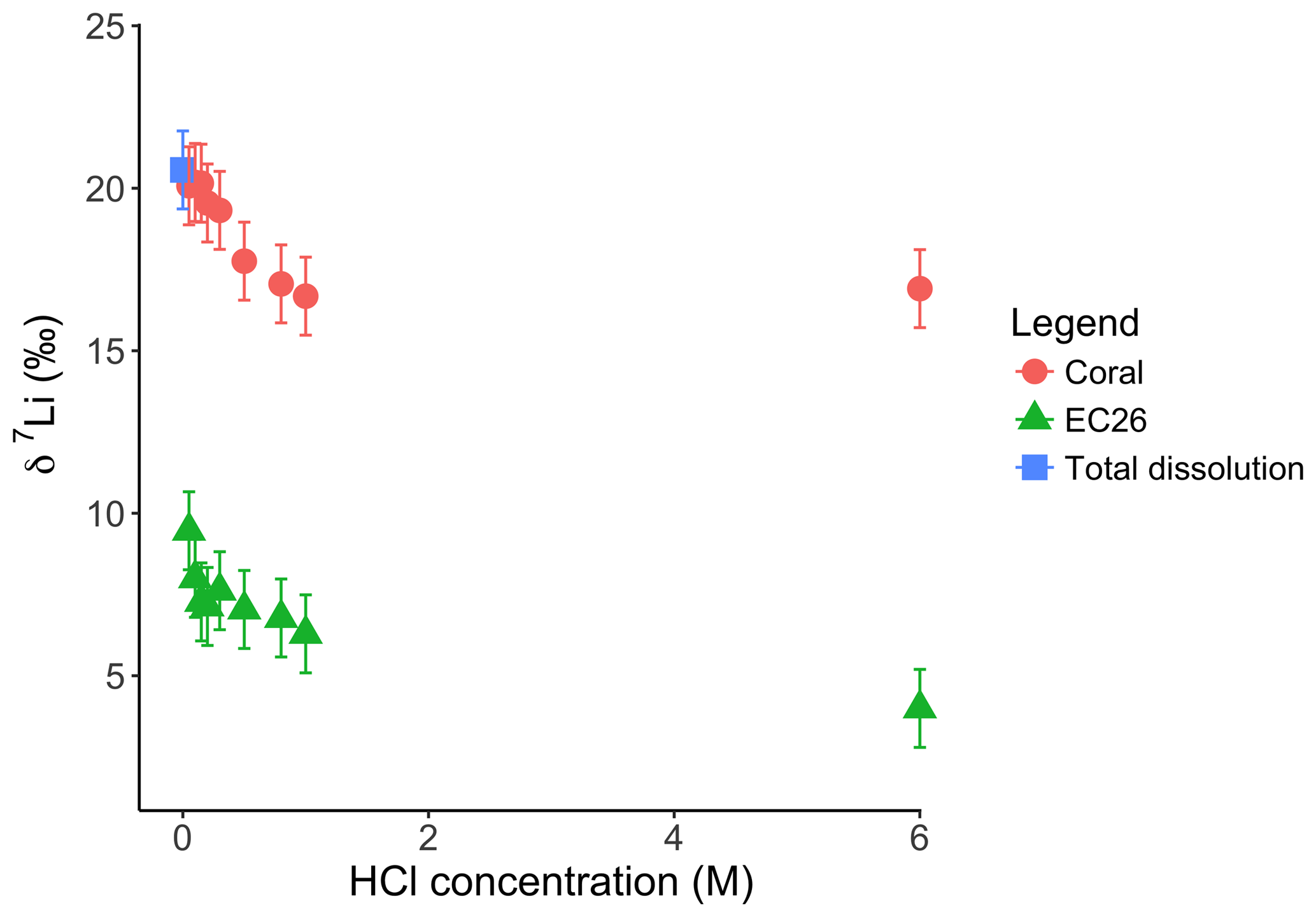
Figure 8Lithium isotope composition of leaching solutions as a function of their HCl concentrations. Triangles and circles represent the δ7Li composition of the solutions used to leach a Neoproterozoic dolostone and modern coral, respectively. The square shows the composition of the coral total dissolution. Both coral and dolostone solutions show similar trends, suggesting the release of silicate-bound Li at higher HCl concentrations. This is surprising as the coral is almost exclusively aragonite, so the release of isotopically light Li is not expected. This also implies that total dissolution in dilute HNO3 does not release isotopically light Li into solution, although no residue was observed during dissolution in dilute HNO3.
The relationship between 103ln αprec-sol and temperature can be used to estimate the temperature dependency for Li isotope fractionation between Ca–Mg carbonate and solution. Using average 103ln αprec-sol values for each reaction temperature, we obtain the following temperature-dependent relationship:
where T is the temperature of precipitation in Kelvin.
Using Eq. (2), the Li isotopic fractionation at 25 ∘C is estimated to be ‰ (1σ) (Fig. 4). Although there is a large error on this estimate, our results suggest that Li isotopic fractionation during dolomite/magnesite precipitation is significantly larger than during calcium carbonate precipitation (Marriott et al., 2004a). This temperature-dependant relationship of Li isotope fractionation in our high-temperature experiments follows the isotope fractionation approach considering equilibrium fractionation (Hoefs, 2015). Furthermore, we see that at high temperature 6Li is preferentially incorporated into the mineral phase over 7Li, which is similar to what has been observed at low temperature for calcium carbonate (Marriott et al., 2004a, b). Finally, despite a wide range of formation conditions between high-temperature synthetic dolomites and low-temperature natural dolomites, Kaczmarek and Sibley (2007) showed that natural and synthetic dolomite form by the same growth mechanisms. This was suggested via the identification of identical growth features in etched natural and synthetic dolomite samples as well as unetched synthetic dolomite. The growth fabrics of high-temperature synthetic dolomite and low-temperature natural dolomite have also been compared by Bullen and Sibley (1984), and these results suggest that high-temperature synthetic dolomites produce fabrics similar to those of naturally grown dolomites. Altogether, these observations suggest that the relationship between Li isotope fractionation and temperature derived from high-temperature experiments may also be valid at low temperatures characteristic of natural environments.
If the temperature of the solution from which dolomite is precipitated is known or can be calculated (e.g. via clumped Δ47 proxy; Winkelstern et al., 2016), the above relationship in combination with the δ7Li of dolostone could potentially be used to determine an estimate for the Li isotopic composition of the precipitating palaeo-solution, e.g. brine or seawater. It is important to note that the applicability to natural systems may be limited to dolomite precipitated inorganically, whereas it has been proposed that bacterial mediation could play a major role in the precipitation of dolomite from natural waters at ambient conditions (Vasconcelos et al., 1995). Bacterial mediation was invoked to solve the “dolomite problem”, i.e. the inability to precipitate dolomite at ambient temperatures; the topic of the origin of dolomite formation is still being hotly debated (Liu et al., 2019; Ahm et al., 2019; Gregg et al., 2015). Other models invoke a primary or secondary (diagenetic) origin for dolomite. Primary marine deposition has been invoked for dolomite formation in many studies, even in Precambrian dolostones (Fairchild and Kennedy, 2007; Rose and Maloof, 2010; Kunzmann et al., 2013; Liu et al., 2014). Thus, where it can be shown that dolomite is of primary origin, its Li isotopic composition could provide valuable information on palaeo-environments.
4.2 Extraction of carbonate-bound Li in dolostones
Leaching of dolostone with solutions of variable HCl concentrations yields δ7Li compositions of the leaching solution that decrease with increasing HCl concentrations, suggesting an increasing contribution of isotopically light Li from detrital silicates, such as clay minerals (Fig. 5a). This hypothesis is supported by a negative relationship between δ7Li values and Li∕Ca ratios of the leaching solutions (Fig. 6), which is similar to results from leaching experiments on the Plenus Marl limestone (Pogge von Strandmann et al., 2013). The Li∕Ca ratio is used instead of Li∕Mg because Mg is also present in silicate minerals. Indeed, δ7Li and Li∕Mg ratios show a positive relationship (Fig. 8b), surprisingly suggesting that dolomite and the detrital component are characterized by high and low Li∕Mg ratios, respectively.
The increasing contribution of silicate minerals with the increasing HCl concentration of the leaching solution is further illustrated by increasing Al∕Mg ratios in the leaching solution (Fig. 5b). The contribution from silicates becomes significant for HCl concentrations >0.5 M. For HCl concentrations <0.8 M, the relationship between Al∕Mg and HCl concentration breaks down (Fig. 5b), indicating that silicates have a negligible role in the composition of the solution. Nevertheless, δ7Li values decrease for HCl concentrations as low as 0.1 M. Thus, we propose that treatment of dolostone with a solution of 0.05 M HCl at room temperature for 60 min is the best compromise between minimizing the contribution of silicates and obtaining enough Li for isotopic analysis.
Leaching experiments were also conducted on a Porites coral from the Holocene to test the proposed protocol, as the δ7Li of modern coral is known (Marriott et al., 2004a; Rollion-Bard et al., 2009). Furthermore, because the aragonitic skeleton of modern corals is generally free of detrital material, we can also test that the chosen leaching protocol yields the same Li isotopic composition in the resulting solution, as with total dissolution of the coral. Total dissolution of the modern coral yields a δ7Li value of 20.6 ‰ (Fig. 8). Leaching solutions with HCl concentrations <0.5 M HCl exhibit δ7Li values within error of that obtained from total dissolution. These values are also consistent with δ7Li compositions between 18.4 ‰ and 19.6 ‰ measured in Porites, and 21 ‰ in Acropora corals (Marriott et al., 2004a). Biomineralization has no major effect on the incorporation of Li in coral or foraminifera as Li has no known biological function. The Li isotopic difference between coral and seawater is −11 ‰ (Marriott et al., 2004a). Therefore, δ7Li values obtained from the total dissolution and for leaching solutions with a HCl concentration <0.5 M would yield a δ7Li composition for modern seawater of 31 ‰, consistent with published values (Misra and Froelich, 2012). Consequently, these results, which are similar to that of Dellinger et al. (2018) suggest that leaching with a 0.05 M HCl solution is appropriate to derive the Li associated with the carbonate fraction only.
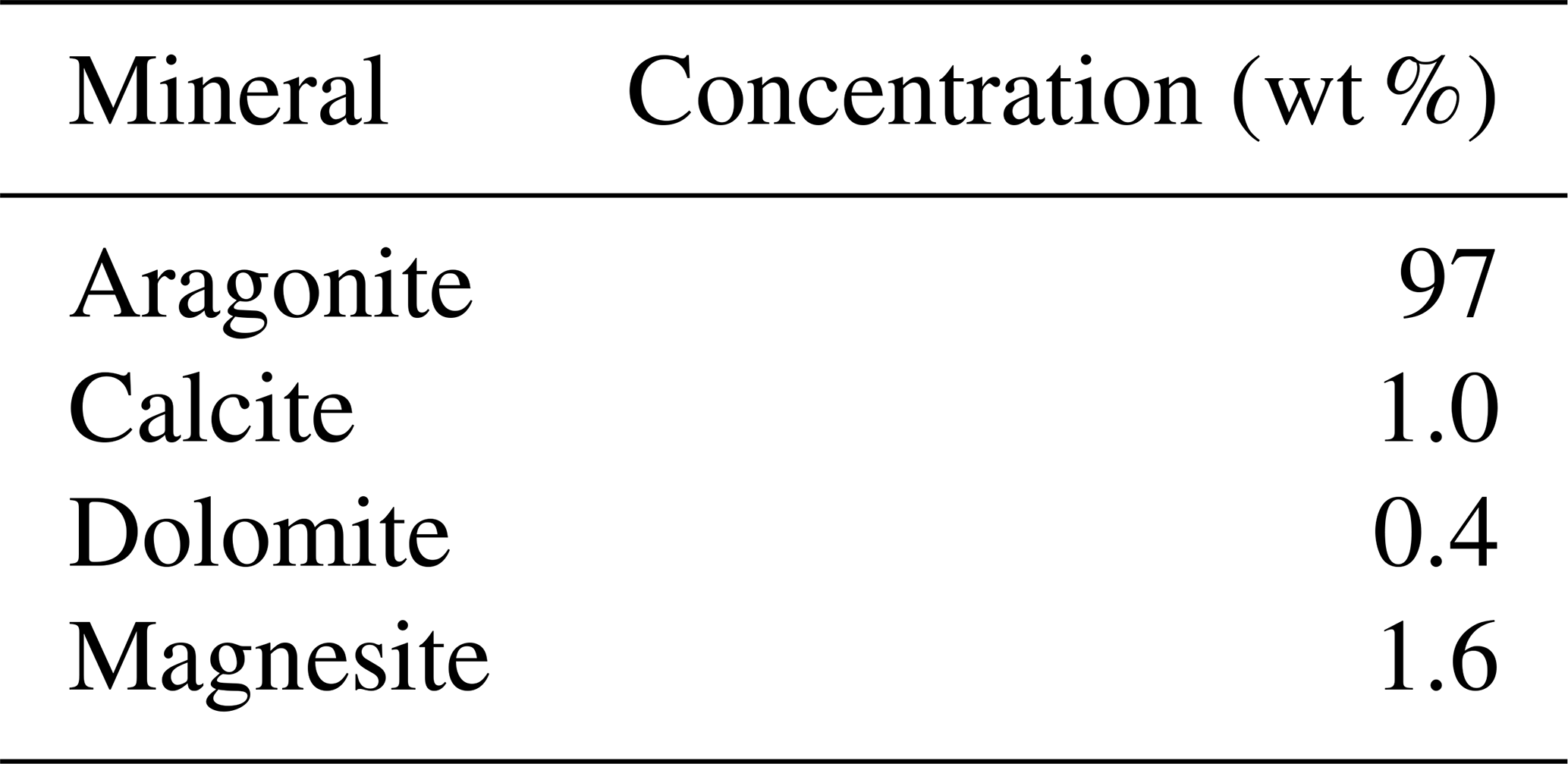
Interestingly both coral and dolostone leaching solutions show a decrease in δ7Li values with increasing HCl concentration. This is surprising as the coral is 97 % aragonite (2 % magnesite and 1 % calcite) so the release of isotopically light Li from silicates is not expected. These results imply that total dissolution in dilute HNO3 does not release isotopically light Li into solution, which could be contained in organic colloids, as no residue was observed. The lack of relationship between δ7Li values and Li∕Ca ratios (Fig. A1) suggests that this isotopically light Li is not bound to silicates (which would have a very different Li∕Ca from aragonite). In the coral, this pool of Li remains unidentified. However, as shown above, leaching with solutions with <0.5 M HCl yield Li isotope compositions expected for a coral in equilibrium with the modern seawater.
Leaching of dolostone with HAc yields δ7Li compositions in the solution similar to that of solutions with a HCl concentration ≤0.1 M (Fig. 7). The δ7Li composition of the 2 % HAc solution is lower (8.37 ‰) than that of the 0.5 % HAc solution, which may suggest a contribution from silicate-bound Li. Thus, treatment of dolostone with a solution of 0.5 % HAc at room temperature for 60 min could be an alternative method to derive carbonate-bound Li.
Precipitation experiments at high temperatures (150, 180 and 220 ∘C) yielded dolomite and magnesite in variable proportions. However, varying mineralogy does not seem to measurably impact Li isotopic fractionation between the carbonate and the solution. The Li isotopic composition of the precipitated solid is isotopically lighter than the reactive solution, which is similar to previous experiments on calcium carbonates (Marriott et al., 2004b, a). The isotope fractionation factor is mainly controlled by temperature, which in turn allows us to calculate the Li isotopic composition of the solution using the δ7Li value of the Ca–Mg carbonate, if the precipitation temperature can be estimated independently (e.g. oxygen or clumped isotope thermometry). Thus, the temperature-dependent relationship in Eq. (2) could be useful for reconstructing δ7Li of palaeo-dolomitizing fluids (i.e. reactive solution) as an approximation based on the Li isotope composition of dolostones in geological records.
Leaching experiments show that it is possible to selectively dissolve the carbonate-bound Li in dolostones by using 0.05 M HCl or 0.5 % HAc at room temperature for 60 min. Leaching of coral with 0.05 M HCl shows that this protocol yields a Li isotope composition for the solution representative of that of the carbonate minerals. Thus, the described protocol allows us to derive the Li isotope composition of the carbonate fraction of dolostones while leaving the Li from any co-present silicates intact.
Combined results from leaching and precipitation experiments show that future studies of Li isotopes in dolostones have considerable potential to further constrain the evolution of the Li isotopic composition of ancient precipitation fluids, including seawater and basinal fluids, and improve our understanding of changes in the Earth's palaeo-environments.
Data will be made available upon request.
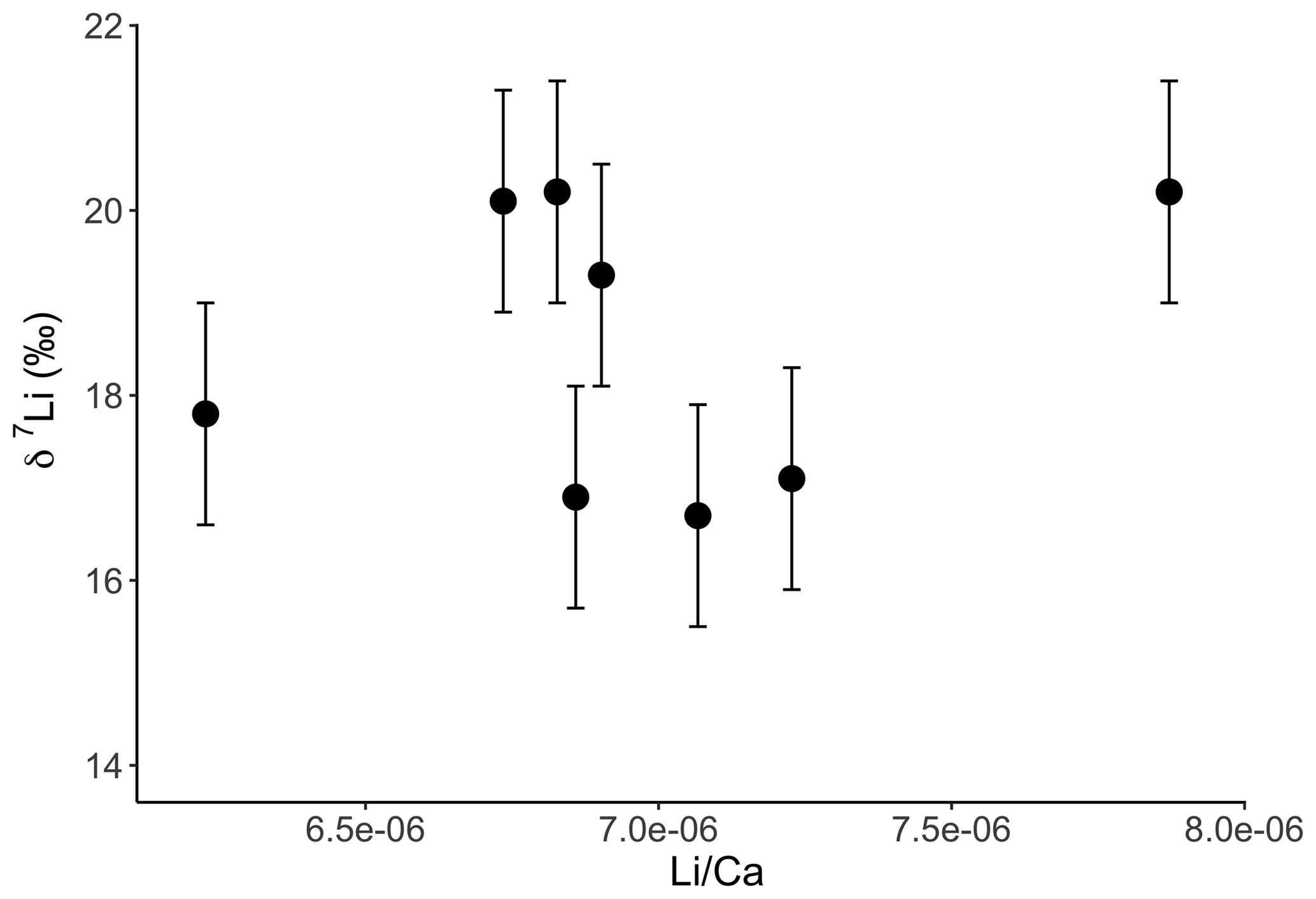
Figure A1Lithium isotope composition of solutions from coral leaching, as a function of their Li∕Ca ratio. In contrast to the dolostone, there is no relationship between δ7Li and Li∕Ca. This could indicate that the isotopically light Li is bound to a fraction with a Li∕Ca similar to that of aragonite. Error bars are within the symbol size, if not shown.
Table A1Column calibration using seawater samples.

Errors are internal analytical uncertainties reported at the 2 σ level. Column C was not used due to the δ7Li value being significantly different from the seawater value.
Table A2Concentrations of lithium in reactive fluids and precipitated solids.
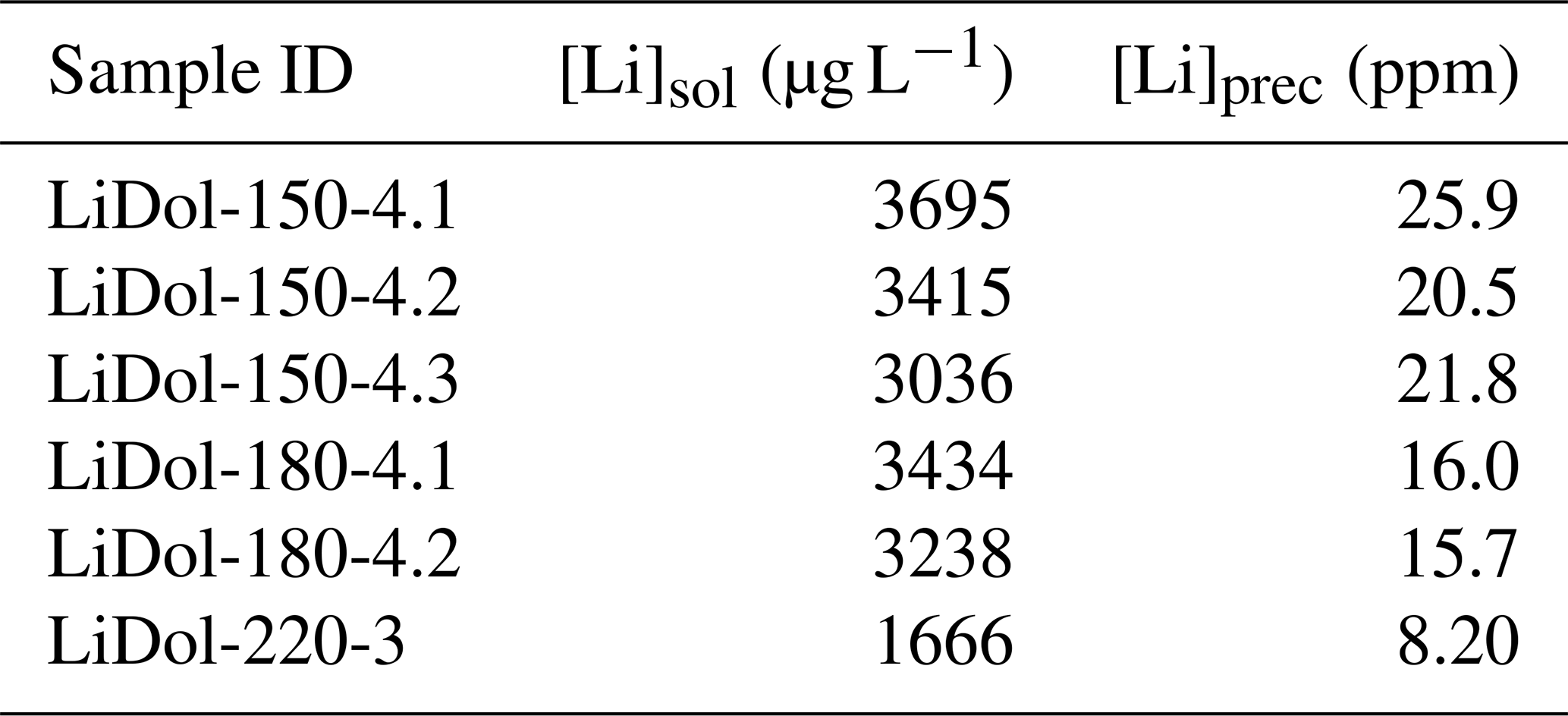
Table A3Mineral concentration of Nuccaleena dolostone (EC26) used in the leaching experiment.
Table A4Mineral concentrations of coral used in the leaching experiment.
HLT, AD, JF and MD designed the project; MD and IJKD conducted the precipitation experiments; HLT conducted the leaching experiments and all other analytical work; HLT and AD wrote the paper; all authors edited the paper.
The authors declare that they have no conflict of interest.
We would like to thank Jasmine Hunter and Helen McGregor (University of Wollongong) for providing the coral samples, Alexander Corrick (University of Adelaide) for help collecting the Nuccaleena dolomite samples and Andre Baldermann (Graz University of Technology) for performing the Li concentration analytics. The laboratory precipitation experiments and fieldwork related to this study was supported by the Base-LiNE Earth project (ITN MC Horizon 2020, grant agreement no. 643084), the Czech Science Foundation (GACR grant no. 17-18120S), the Australian Government Research Training Program and the ARC Linkage Project LP160101353.
This paper was edited by Yves Godderis and reviewed by Yves Godderis and one anonymous referee.
Ahm, A.-S. C., Maloof, A. C., Macdonald, F. A., Hoffman, P. F., Bjerrum, C. J., Bold, U., Rose, C. V., Strauss, J. V., and Higgins, J. A.: An early diagenetic deglacial origin for basal Ediacaran “cap dolostones”, Earth Planet. Sc. Lett., 506, 292–307, https://doi.org/10.1016/J.EPSL.2018.10.046, 2019. a
Arvidson, R. S. and Mackenzie, F. T.: The dolomite problem; control of precipitation kinetics by temperature and saturation state, Am. J. Sci., 299, 257–288, 1999. a
Balter, V. and Vigier, N.: Natural variations of lithium isotopes in a mammalian model, Metallomics, 6, 582–586, https://doi.org/10.1039/C3MT00295K, 2014. a
Bastian, L., Vigier, N., Reynaud, S., Kerros, M.-E., Revel, M., and Bayon, G.: Lithium Isotope Composition of Marine Biogenic Carbonates and Related Reference Materials, Geostand. Geoanal. Res., 42, 403–415, https://doi.org/10.1111/ggr.12218, 2018. a
Bullen, S. B. and Sibley, D. F.: Dolomite selectivity and mimic replacement, Geology, 12, 655–658, https://doi.org/10.1130/0091-7613(1984)12<655:DSAMR>2.0.CO;2, 1984. a
Burns, S. J., Mckenzie, J. A., and Vasconcelos, C.: Dolomite formation and biogeochemical cycles in the Phanerozoic, Sedimentology, 47, 49–61, 2000. a
Carignan, J., Vigier, N., and Millot, R.: Three secondary reference materials for lithium isotope measurements: Li7-N, Li6-N and LiCl-N solutions, Geostand. Geoanal. Res., 31, 7–12, 2007. a, b
Dellinger, M., West, A. J., Paris, G., Adkins, J. F., von Strandmann, P. A. P., Ullmann, C. V., Eagle, R. A., Freitas, P., Bagard, M.-L., and Ries, J. B.: The Li isotope composition of marine biogenic carbonates: Patterns and Mechanisms, Geochim. Cosmochim. Ac., 236, 315–335, 2018. a
Fairchild, I. J. and Kennedy, M. J.: Neoproterozoic glaciation in the Earth System, J. Geol. Soc., 164, 895–921, https://doi.org/10.1144/0016-76492006-191, 2007. a
Flesch, G. D., Anderson, A. R., and Svec, H. J.: A secondary isotopic standard for 6Li∕7Li determinations, Int. J. Mass Spectrom., 12, 265–272, 1973. a
Geske, A., Zorlu, J., Richter, D., Buhl, D., Niedermayr, A., and Immenhauser, A.: Impact of diagenesis and low grade metamorphosis on isotope (δ26Mg, δ13C, δ18O and 87Sr∕86Sr) and elemental (Ca, Mg, Mn, Fe and Sr) signatures of Triassic sabkha dolomites, Chem. Geol., 332, 45–64, 2012. a
Gregg, J. M., Bish, D. L., Kaczmarek, S. E., and Machel, H. G.: Mineralogy, nucleation and growth of dolomite in the laboratory and sedimentary environment: A review, Sedimentology, 62, 1749–1769, https://doi.org/10.1111/sed.12202, 2015. a, b
Hathorne, E. C. and James, R. H.: Temporal record of lithium in seawater: A tracer for silicate weathering?, Earth Planet. Sc. Lett., 246, 393–406, 2006. a
Hoefs, J.: Theoretical and Experimental Principles, in: Stable Isotope Geochemistry, Springer International Publishing, Cham, 1–46, https://doi.org/10.1007/978-3-319-19716-6_1, 2015. a
James, R. H. and Palmer, M. R.: The lithium isotope composition of international rock standards, Chem. Geol., 166, 319–326, 2000. a
Kaczmarek, S. E. and Sibley, D. F.: A Comparison of Nanometer-Scale Growth and Dissolution Features on Natural and Synthetic Dolomite Crystals: Implications for the Origin of Dolomite, J. Sediment. Res., 77, 424–432, https://doi.org/10.2110/jsr.2007.035, 2007. a
Yamaji, K., Makita, Y., Watanabe, H., Sonoda, A., Kanoh, H., Hirotsu, T., and Ooi, K.: Theoretical Estimation of Lithium Isotopic Reduced Partition Function Ratio for Lithium Ions in Aqueous Solution, J. Phys. Chem. A, 105, 602–613, https://doi.org/10.1021/JP001303I, 2001. a
Kunzmann, M., Halverson, G. P., Sossi, P. A., Raub, T. D., Payne, J. L., and Kirby, J.: Zn isotope evidence for immediate resumption of primary productivity after snowball Earth, Geology, 41, 27–30, https://doi.org/10.1130/g33422.1, 2013. a
Land, L. S.: Failure to precipitate dolomite at 25 degrees C from dilute solution despite 1000-fold oversaturation after 32 years, Aquat. Geochem., 4, 361–368, https://doi.org/10.1023/A:1009688315854, 1998. a
Lechler, M., Pogge von Strandmann, P. A. E., Jenkyns, H. C., Prosser, G., and Parente, M.: Lithium-isotope evidence for enhanced silicate weathering during OAE 1a (Early Aptian Selli event), Earth Planet. Sc. Lett., 432, 210–222, 2015. a
Li, D. D., Jacobson, A. D., and McInerney, D. J.: A reactive-transport model for examining tectonic and climatic controls on chemical weathering and atmospheric CO2 consumption in granitic regolith, Chem. Geol., 365, 30–42, https://doi.org/10.1016/j.chemgeo.2013.11.028, 2014. a
Liu, C., Wang, Z., Raub, T. D., Macdonald, F. A., and Evans, D. A. D.: Neoproterozoic cap-dolostone deposition in stratified glacial meltwater plume, Earth Planet. Sc. Lett., 404, 22–32, https://doi.org/10.1016/j.epsl.2014.06.039, 2014. a
Liu, D., Xu, Y., Papineau, D., Yu, N., Fan, Q., Qiu, X., and Wang, H.: Experimental evidence for abiotic formation of low-temperature proto-dolomite facilitated by clay minerals, Geochim. Cosmochim. Ac., 247, 83–95, https://doi.org/10.1016/J.GCA.2018.12.036, 2019. a
Marriott, C. S., Henderson, G. M., Belshaw, N. S., and Tudhope, A. W.: Temperature dependence of δ7Li, δ44Ca and Li∕Ca during growth of calcium carbonate, Earth Planet. Sc. Lett., 222, 615–624, 2004a. a, b, c, d, e, f, g, h, i, j, k
Marriott, C. S., Henderson, G. M., Crompton, R., Staubwasser, M., and Shaw, S.: Effect of mineralogy, salinity, and temperature on Li∕Ca and Li isotope composition of calcium carbonate, Chem. Geol., 212, 5–15, 2004b. a, b, c, d, e
Misra, S. and Froelich, P. N.: Lithium isotope history of Cenozoic seawater: Changes in silicate weathering and reverse weathering, Science, 335, 818–823, https://doi.org/10.1126/science.1214697, 2012. a, b
Pistiner, J. S. and Henderson, G. M.: Lithium-isotope fractionation during continental weathering processes, Earth Planet. Sc. Lett., 214, 327–339, https://doi.org/10.1016/s0012-821x(03)00348-0, 2003. a
Pogge von Strandmann, P. A. E., Jenkyns, H. C., and Woodfine, R. G.: Lithium isotope evidence for enhanced weathering during Oceanic Anoxic Event 2, Nat. Geosci., 6, 668–672, 2013. a, b, c
Pogge von Strandmann, P. A. E., Desrochers, A., Murphy, M. J., Finlay, A. J., Selby, D., and Lenton, T. M.: Global climate stabilisation by chemical weathering during the Hirnantian glaciation, Geochem. Perspect. Lett., 3, 230–237, https://doi.org/10.7185/geochemlet.1726, 2017. a
Rollion-Bard, C., Vigier, N., Meibom, A., Blamart, D., Reynaud, S., Rodolfo-Metalpa, R., Martin, S., and Gattuso, J.-P.: Effect of environmental conditions and skeletal ultrastructure on the Li isotopic composition of scleractinian corals, Earth Planet. Sc. Lett., 286, 63–70, https://doi.org/10.1016/j.epsl.2009.06.015, 2009. a
Rose, C. V. and Maloof, A. C.: Testing models for post-glacial “cap dolostone” deposition: Nuccaleena Formation, South Australia, Earth Planet. Sc. Lett., 296, 165–180, https://doi.org/10.1016/j.epsl.2010.03.031, 2010. a
Teng, F. Z., Dauphas, N., and Helz, R. T.: Iron Isotope Fractionation During Magmatic Differentiation in Kilauea Iki Lava Lake, Science, 320, 1620–1622, https://doi.org/10.1126/science.1157166, 2008. a
Vasconcelos, C., McKenzie, J. A., Bernasconi, S., Grujic, D., and Tiens, A. J.: Microbial mediation as a possible mechanism for natural dolomite formation at low temperatures, Nature, 377, 220–222, https://doi.org/10.1038/377220a0, 1995. a
Vigier, N. and Goddéris, Y.: A new approach for modeling Cenozoic oceanic lithium isotope paleo-variations: the key role of climate, Clim. Past, 11, 635–645, https://doi.org/10.5194/cp-11-635-2015, 2015. a
Wanner, C., Sonnenthal, E. L., and Liu, X.-M.: Seawater δ7Li: A direct proxy for global CO2 consumption by continental silicate weathering?, Chem. Geol., 381, 154–167, https://doi.org/10.1016/j.chemgeo.2014.05.005,2014. a
Winkelstern, I. Z., Kaczmarek, S. E., Lohmann, K. C., and Humphrey, J. D.: Calibration of dolomite clumped isotope thermometry, Chem. Geol., 443, 32–38, https://doi.org/10.1016/j.chemgeo.2016.09.021, 2016. a